Crux Frequently Asked Questions
- What is the difference between Tide and Comet?
-
Tide converts your FASTA file to an index (either as part
of
tide-searchor by usingtide-index), whereas Comet does searching directly from the FASTA file. The indexing step allows Tide to pre-compute many parts of the search procedure, thereby making the search faster. - Comet is multi-threaded, whereas Tide runs on a single thread. On multi-threaded CPUs, this feature can offset the speed difference between Comet and Tide.
- For each match, Comet reports the total number of candidate peptides, whereas Tide reports the number of distinct candidate peptides.
- Comet provides an option to report an E-value in addition to the raw XCorr score, whereas Tide provides an option to report a p-value. The methods for computing these two statistical confidence measures are quite different.
- Some options are available only in Comet (various options related to different types of theoretical fragment ions, max_fragment_charge, nucleotide_reading_frame, etc.) whereas others are only available in Tide (e.g., keep-terminal-aminos).
- What operating systems does Crux work with?
- How does Crux compute the masses of peptides and peptide fragments?
-
Each amino acid can be characterized either by its monoisotopic mass,
which is the mass of the most abundant isotopic form of that amino acid,
or the average mass, which is a weighted average of the masses of all
the isotopic forms. By default, Crux uses average mass to calculate
peptide masses, though this behavior can be controlled by the
isotopic-massoption totide-indexand themass_type_parentoption tocomet. For fragments,tide-searchalways uses the monoisotopic mass, whereascometallows selection viamass_type_fragment. - The neutral mass of a peptide is not the sum of the masses of its amino acids. The N-terminus and C-terminus of the peptide together contribute an additional water molecule, whose mass is either 18.010564684 Da (monoisotopic) or 18.0153 Da (average).
- A charged peptide has an additional approximately 1 Da mass for each charge, corresponding to the mass of a hydrogen atom. The exact mass to be added depends on whether we are using the monoisotopic mass (1.007825035) or average mass (1.00794).
- When a peptide fragments, each b-ion will have a mass equal to the sum of its amino acids plus one hydrogen for each charge on the fragment, while each y-ion will have a mass equal to the sum of its amino acids plus water plus one hydrogen for each charge on the fragment.
-
By default, both Tide and Comet add a static modification of
57.021464 Da to all cysteines. This is because in most protein
preparation protocols the peptides are alkylated with iodoacetamide,
resulting in carbamidomethylation of cysteine. The alkylation step
prevents the reformation of disulfide bonds. Other alykylation
reagents may be used, in which case the appropriate mass shift can be
specified with the
C=[mass]option totide-indexor theadd_C_cysteineoption tocomet. - How does Crux assign fragment masses to bins prior to computing the XCorr score?
- How does Crux estimate the possible charge states of the peptides when the information is not provided within the spectra file?
- How does Crux create peptides from the given set of proteins in the database?
- How does Crux select candidate peptides from the database?
-
If the
precursor-window-typeis set tomz, then the window is calculated as spectrum precursor m/z ±precursor-windowand the resulting range is converted to mass using the charge state with the formula:
Mass=m/z * charge - MASS_PROTON * charge, where MASS_PROTON=1.00727646677 -
If the
precursor-window-typeis set tomass, then the window is defined as the precursor mass ±precursor-window. -
If the
precursor-window-typeis set toppm(parts per million), then the window spans from the precursor mass / (1.0 + window * 1e-6) to precursor mass / (1.0 - window * 1e-6). - How can I search my ITRAQ data?
- What happens if I decrease the size of the precursor window during searching?
-
How can I run many jobs in parallel?
In combination with GNU parallel, parallelization of Crux can be achieved rather painlessly. For example, to run tide-search on a set of spectrum files, you can just get your spectra files into a filelist:
find . -name "*.mzXML" > file.list
Then, with a bit of bash magic, run parallel as follows (assuming there are 100 files in file.list):
parallel --xapply crux tide-search <options> --fileroot {1} {2} database.db ::: $(seq 1 100) ::: $(cat file.list)Thanks to Ben Temperton for this answer.
- How can I contribute to Crux?
- Where does the name "Crux" come from?
The differences are as follows:
Overall, for a given set of spectra, the XCorr scores computed by Comet and by Tide should be quite similar to one another, assuming that the various search parameters are set similarly between the two algorithms. Below is a table summarizing the correspondence between the two sets of parameters. The specified values will yield nearly identical XCorr scores from the two search engines.
| Tide parameter | Tide value | Comet parameter | Comet value |
| enzyme | trypsin | search_enzyme_number | 1 |
| digestion | full-digest | num_enzyme_termini | 2 |
| missed-cleavages | 0 | allowed_missed_cleavage | 0 |
| min-peaks | 10 | minimum_peaks | 10 |
| precursor-window | 3 | peptide_mass_tolerance | 3 |
| precursor-window-type | mass | peptide_mass_units | 0 |
| monoisotopic-precursor | T | mass_type_parent | 1 |
| fragment-mass | mono | mass_type_fragment | 1 |
| decoy-format | peptide-reverse | N/A | |
| num-decoys-per-target | 1 | N/A | |
| keep-terminal-aminos | C | N/A | |
| concat | T | decoy_search | 1 |
| top-match | 5 | num_results, num_output_lines | 6, 5 |
| remove-precursor-peak | T | remove_precursor_peak | 1 |
| remove-precursor-tolerance | 15 | remove_precursor_tolerance | 15 |
| use-flanking-peaks | F | theoretical_fragment_ions | 1 |
| use-neutral-loss-peaks | F | use_NL_ions | F |
| fragment-bin-width | 1.0005079 | fragment_bin_tol | 1.0005079 |
| mz-bin-offset | 0.4 | fragment_bin_offset | 0.4 |
| min-mass, max-mass | 200, 7200 | digest_mass_range | 200 7200 |
| N/A | max_fragment_charge | 2 |
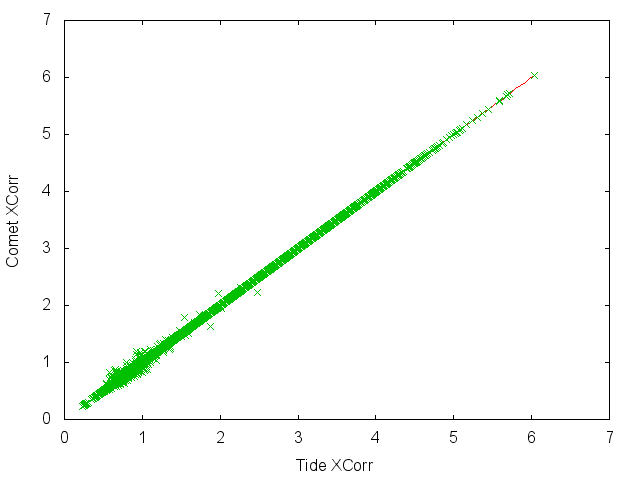
Here is a scatter plot of XCorr scores from a search run with the parameters listed above:
Crux is written in C++, so in principle it should work on virtually any modern operating system. We provide pre-compiled binaries for Linux, MacOS and Windows.
The XCorr score is essentially a dot product between a preprocessed
form of the observed spectrum and the theoretical spectrum derived
from the candidate peptide. In order to compute this dot product, the
masses of the fragments in both spectra are assigned to discrete mass
bins. This can be viewed as a form of rounding, but with more control
over the discrete masses. Two parameters, mz-bin-width and
mz-bin-offset, control the size and location of the bins,
and are used to convert fragment masses according to this formula:
binned mass = floor( ( original mass / bin-width ) + 1.0 - bin-offset )
The default values bin-width=1.0005079
and bin-offset=0.40 are suitable for most low-resolution
datasets.
Crux calculates the ratio of two values: (1) the sum of the intensities from the peaks in the fragmentation spectrum whose m/z is greater than the precursor m/z versus (2) the sum of the peaks whose m/z is smaller than the precursor. If this ratio is greater than or equal to a calculated threshold based upon the location of the precursor m/z and the max m/z in the spectrum, Crux then assigns both +2 and +3 as possible charge states to the spectrum. Otherwise, Crux assigns +1 as the charge state. The algorithm is based upon the observation that fragmentation spectra collected from +1 peptides should have no peaks above the precursor m/z. In contrast, a peptide of charge state greater than +1 can generate fragment ions of lower charge whose m/z is greater than the precursor m/z, thus indicating a multiply charged precursor.
Note that, in addition, comet includes
a precursor_charge parameter. If the first number in
this parameter is set to 0, i.e., precursor_charge = 0 0,
then the charge state rule above is applied. However, if a user
specifies a precursor charge range, i.e., precursor_charge = 1
5, then Comet will search every spectrum through this range of
assumed charge states for every spectrum whose precursor charge
is unknown.
The options enzyme (for tide-index)
and search_enzyme_number (for comet) define
the enzymatic cleavage rules. When enzyme=no-enzyme
or search_enzyme_number=0, then any subsequence of a
protein may be considered as a candidate peptide. For other values of
these parameters, the residues at the termini of the protein
subsequence must follow specific rules. For example, trypsin requires
that the preceeding residue must be an R or K and the following
residue may not be a P.
When digestion=full-digest (for tide-index)
or num_enzyme_termini=2 (for comet), then
these rules must be true at both ends of the peptide. When it
is digestion=partial-digest
or num_enzyme_termini=1, then the rules must be true at
at least one of the ends. The missed-cleavages
and allowed_missed_cleavages parameters control the
maximum number of cleavage sites that may lie within the peptide
sequence.
Note that if enzyme=no-enzyme
or search_enzyme_number=0, then
the digestion, num_enzyme_termini, missed-cleavages
and allowed_missed_cleaveages parameters are not used.
The Crux search tools (tide-index and comet)
select candidate peptides for each spectrum based on its precursor
singly charged mass (m+h) or the mass-to-charge (m/z) and an assumed
charge state (specified in the input file). If the m+h and charge is
provided in the input file (e.g., from the Z lines of an MS2 file),
then the precursor mass is calculated from the m+h minus the mass of a
proton. Otherwise, the precursor mass is calculated from the precursor
m/z and an assumed charge. A mass window is defined in one of three
ways based on the
precursor-window-type and precursor-window
options.
Candidate peptides are those whose mass falls within the defined window. The peptide mass is computed as the sum of the average amino acid masses plus 18 Da for the terminal OH group. Candidate peptides can also be constrained by minimum and maximum allowed length.
ITRAQ and other tagged seaches can be done by using terninal modifications. For example, if your tag has a mass of 304.199040, add these lines to your parameter file:
nmod=304.199040:-1 cmod=304.199040:-1
Crux will then generate peptides with this modification (+304.199904) on the n-terminus, the c-terminus and on both termini. The -1 in the option means to add the modification regardless of where the peptide terminus lies relative to the rest of the protein.
Reducing the precursor mass tolerance has two main benefits. One is
reduced search time, and the other is improved statistical power to
detect matches. With a smaller window, fewer candidates are tested
against a spectrum. As a result, the statistical confidence measure
calculated after multiple testing correction will be more significant.
Of course, the flipside is that if you make the precursor window too
small, then you may end up throwing out correct identifications.
Control over the size of the window is provided by the using
the precursor-window and
precursor-window-type options.
Patches implementing new features can be emailed to the development team at crux-support@uw.edu for review and inclusion in subsequent releases of Crux.
Thin air. The name is not an acronym or a reference to anything in particular.