 Download Crux Toolkit
Download Crux Toolkit
|
|
Crux is a software toolkit for tandem mass spectrometry analysis. If you use Crux in your research, please cite
Christopher Y. Park, Aaron A. Klammer, Lukas Käll, Michael J. MacCoss and William Stafford Noble. "Rapid and accurate peptide identification from tandem mass spectra." Journal of Proteome Research. 7(7):3022-3027, 2008.
For a more up-to-date description of Crux, please read
Sean McIlwain, Kaipo Tamura, Attila Kertesz-Farkas, Charles E. Grant, Benjamin Diament, Barbara Frewen, J. Jeffry Howbert, Michael R. Hoopmann, Lukas Käll, Jimmy K. Eng, Michael J. MacCoss and William Stafford Noble. "Crux: rapid open source protein tandem mass spectrometry analysis." Journal of Proteome Research. 13(10):4488-4491, 2014.
| Primary commands | |
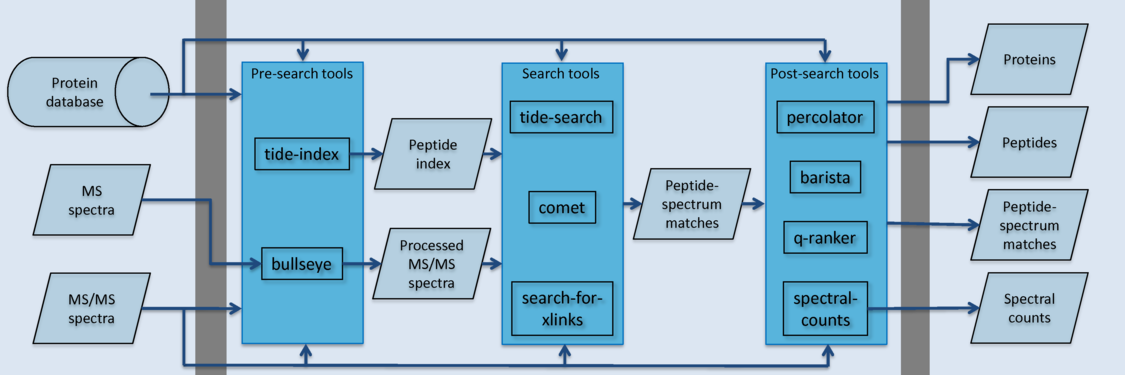
| bullseye | Assign high resolution precursor m/z values to MS/MS data using the Hardklör algorithm. |
| tide-index |
Create an index of all peptides in a fasta file, for use in subsequent
calls to tide-search. |
| tide-search |
Search a collection of spectra against a sequence database, provided
either as a FASTA file or an index, returning a collection of
peptide-spectrum matches (PSMs). This is a fast search engine, but it
runs most quickly if provided with a peptide index built
with tide-index. |
| cascade-search | An iterative procedure for incorporating information about peptide groups into the database search and confidence estimation procedure. |
| comet | Search a collection of spectra against a sequence database, returning a collection of PSMs. This search engine runs directly on a protein database in FASTA format. |
| percolator | Re-rank and assign confidence estimates to a collection of PSMs using the Percolator algorithm. Optionally, also produce protein rankings using the Fido algorithm. |
| q-ranker | Re-rank a collection of PSMs using the Q-ranker algorithm. |
| barista | Rank PSMs, peptides and proteins, assigning a confidence measure to each identification. |
| search-for-xlinks | Search a collection of spectra against a sequence database, finding cross-linked peptide matches. |
| spectral-counts | Quantify peptides or proteins using one of three spectral counting methods. |
| pipeline | Given one or more sets of tandem mass spectra as well as a protein database, this command runs a series of Crux tools and reports all of the results in a single output directory. |
| Utilities | |
| assign-confidence | Assign statistical confidence measures to each PSM in a given set. |
| generate-peptides | Extract from a given set of protein sequences a list of target and decoy peptides fitting the specified criteria. |
| get-ms2-spectrum |
Extract one or more fragmentation spectra, specified by scan number, from
an MS2 file. |
| hardklör | Identify isotopic distributions from high-resolution mass spectra. |
| make-pin | Given a set of search results files, generate a pin file for input to
crux percolator |
| predict-peptide-ions | Given a peptide and a charge state, predict the m/z values of the resulting fragment ions. |
| print-processed-spectra | Process spectra as for scoring xcorr and print the results to a file. |
| psm-convert | Convert a file containing peptide-spectrum matches (PSMs) from one format to another. |
| subtract-index | Subtract one index file from another, assuming both were generated by tide-index. |
| xlink-assign-ions | Given a spectrum and a pair of cross-linked peptides, assign theoretical ion type labels to peaks in the observed spectrum. |
| xlink-score-spectrum | Takes a defined cross-linked peptide, a spectra file, and a scan number, and will calculate the XCorr score a number of different ways. |
| version | Print the Crux version number to standard output, then exit. |
| Utilities for processing tab-delimited text files | |
| extract-columns | Print specified columns from a tab-delimited file. |
| extract-rows | Print specified rows from a tab-delimited file. |
| stat-column | Collect summary statistics from a column in a tab-delimited file. |
| sort-by-column | Sort a tab-delimited file by a column. |
File formats
| bullseye | tide-index | tide-search | comet | percolator | q-ranker | barista | search-for-xlinks | spectral-counts | |
| MS1 | in | ||||||||
| MS2 (sample) / CMS2 | in/out | in | in | in | in | in | in | in | |
| MGF | in/out | in | in | in | in | in | in | ||
| mzML, mzXML | in | in | in | in | in | in | in | in | |
| Thermo .raw files (Windows only) | in | in | in | in | in | in | in | ||
| Other proprietary vendor formats (Windows only) | in | in | in | in | in | ||||
| FASTA (sample) | in | in | in | in | in | ||||
| database index | out | in | |||||||
| Tab-delimited text (sample) | out | out | in/out | in/out | in/out | out | in/out | ||
| pepXML | out | out | in/out | out | out | in | |||
| PIN | out | in | |||||||
| POUT | out | ||||||||
| mzIdentML | out | out | out | in | |||||
| SQT | out | out | in | in | in | in | |||
| Feature file | in/out | out | |||||||
| Barista XML | out | ||||||||
| Crux parameters (sample) | in/out | in/out | in/out | in/out | in/out | in/out | in/out | in/out | in/out |
Tutorials
- Installation
- Getting started with Crux
- Running a simple search using Tide and Percolator
- Customization and search options
- Using spectral-counts
Important links
- Crux frequently asked questions
- Glossary of terminology
- A graphical user interface for the Crux search tools, as well as a variety of other search tools, is available through SearchGUI. The sister program, PeptideShaker provides a way to combine the results of multiple search engines in the analysis of a single data set.
- Crux Google Scholar profile
- To receive announcements of new versions, sign up for the Crux users mailing list.
- For support, and to discuss Crux with other users and the developers, sign up for the Crux users Google group.
- Follow the Crux project on Sourceforge. The "Issues" list on Sourceforge includes known bugs as well as planned new features.
-
Access the most recent source code from the Crux main development
trunk using the following Subversion command:
svn checkout svn://svn.code.sf.net/p/cruxtoolkit/code/crux/trunk crux-trunk - Release notes.
- Crux is covered by an Apache license.
The original version of Crux was written by Chris Park and Aaron Klammer under the supervision of Prof. Michael MacCoss and Prof. William Stafford Noble in the Department of Genome Sciences at the University of Washington, Seattle. The complete list of contributors can be found here.
Maintenance and development of Crux is funded by the National Institutes of Health awards R01 GM096306 and P41 GM103533.
Please send comments and questions to crux-developers@uw.edu or crux-users@googlegroups.com.